
Cystic fibrosis is a rare disease caused by a genetic mutation that can be passed down from parent to child. Although there is no known cure, there are many treatments and medications for cystic fibrosis that can help reduce the effects and symptoms of the disease.
The most prominent symptom of cystic fibrosis is abnormal, thick mucus that builds up in the lungs and other organs. This causes blockages and infections that can cause a variety of serious complications, including lung disease, liver disease, and gastrointestinal (GI) problems.
Many people with cystic fibrosis fail to grow and thrive as children and experience frequent infections and organ damage throughout their lives. It is a deadly disease, and the median age of survival for people with cystic fibrosis is around 40 years old.
{{cta('fa8abc2a-1e88-4fa3-82fd-1cb5b9ed43b2','justifycenter')}}
Cystic fibrosis is complex and affects many different parts of the body, including the lungs, pancreas, liver, and intestines, in different ways. People with cystic fibrosis have to take a variety of medications, adhere to special diets, and do daily respiratory exercises to combat serious problems like malnutrition and difficulty breathing.
Symptoms of cystic fibrosis usually show up very early in life, and most people are diagnosed by the time they turn two. The disease requires daily, life-long management to keep uncomfortable symptoms at bay and prevent serious damage to the respiratory tract, digestive system, and other organs.
Cystic fibrosis is a life-altering condition that requires intensive treatment to manage. However, recent advancements in cystic fibrosis treatment have made huge steps toward improving patents' lifespans and quality of life. With proper medication, strict daily treatment regimens, and close monitoring from doctors and specialists, people with cystic fibrosis are now able to live better and longer than ever before.
In this article we will introduce you to the signs and symptoms of cystic fibrosis and how the disease effects many different parts of the body. You'll learn about cystic fibrosis treatments, potential complications, and how the disease gets passed down through generations.
What is Cystic Fibrosis?
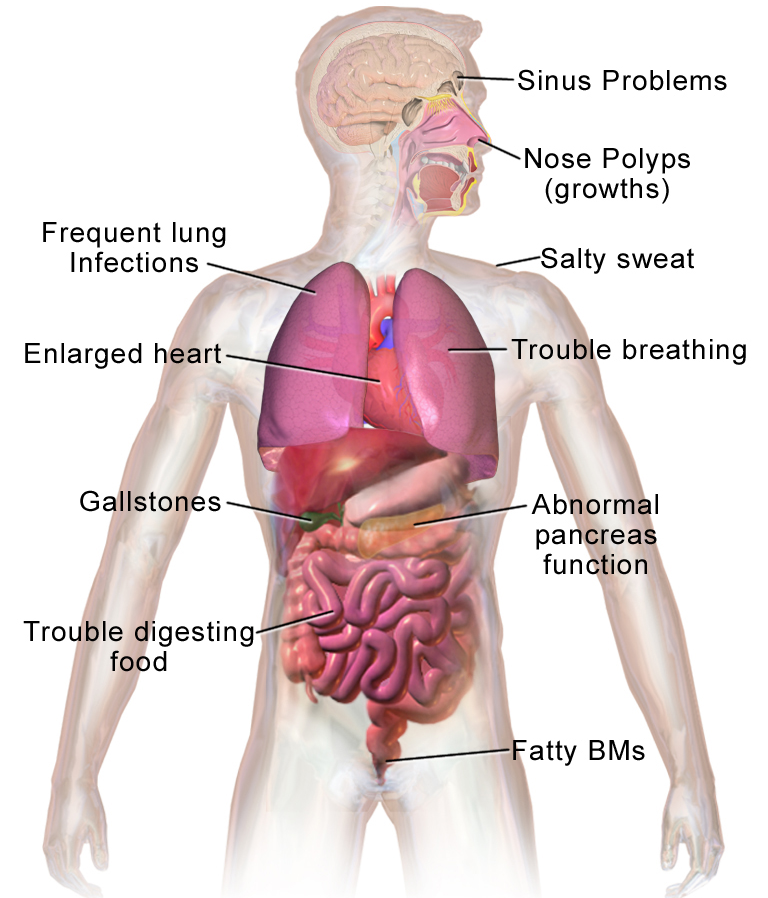
Cystic fibrosis can be difficult to understand because it comes with such a wide range of varied symptoms and complications. It doesn't just damage lungs or one type of tissue; it can affect many different parts of the body.
All people with cystic fibrosis have a mutation in a gene that controls a vital protein called the cystic fibrosis trans-membrane regulator (CFTR). This protein is an important part of membranes found all throughout the body, specifically in tissues that secrete mucus like the lungs and intestines.
These tissues are responsible for producing thin, watery mucus to coat the insides of the lungs and other organs. This lubricates and protects the delicate inner membranes, but can be problematic if it gets too dry and thick.
A normal, functioning CFTR protein is necessary for the body to be able to secrete normal mucus as well as thin, flowing fluids that other organs, like the pancreas, produce. Unfortunately, people who have cystic fibrosis don't have enough of this protein working in their bodies.
That is because a genetic defect causes one of two things to happen:
-
The body makes an abnormal version of the CFTR protein that doesn't function properly.
- The body can't make enough of the CFTR protein.
In either case, this results in mucus and other secretions that don't function properly. Instead of producing thin, flowing mucus, people with cystic fibrosis produce dry, thick mucus that sticks to insides of their lungs and other organs.
This thickened mucus can't do its job and instead clogs up organs and provides an ideal spot for bacteria to grow. It also has specific effects on the liver, gut, and pancreas that disrupt their normal function and can lead to serious disease.
How Cystic Fibrosis Affects Your Body
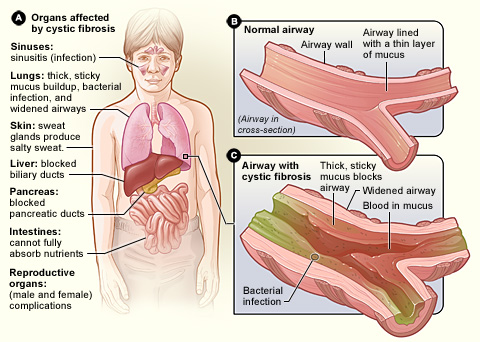
Cystic fibrosis symptoms vary from person to person,and there are multiple types of CFTR gene mutations that can cause the disease. Every case of cystic fibrosis is different, and some are more severe than others.
But one thing all people with cystic fibrosis share is a buildup of excess mucus and phlegm in their bodies, especially in their lungs and digestive tract. This is because the defective CFTR protein cannot properly regulate the water content of the body's secretions, leading to mucus that's extra thick and sticky, instead of thin and free-flowing.
This has a devastating effect on many parts of the body, and we'll go through them here one by one.
Lungs
When you have cystic fibrosis, the thickened mucus in your lungs dries up easily and clogs up your airways. This thick, sticky mucus is difficult to clear out and sits in place, inviting bacteria to build up in the airways and infect the lungs.
The mucus buildup blocks air from flowing through your lungs easily, causing breathlessness, coughing, and reduced lung function. One of the most important parts of cystic fibrosis treatment is clearing mucus out of the airways and keeping the lungs as healthy as possible.
People with cystic fibrosis often suffer from frequent or chronic lung infections that can cause a chronic, phlegmy cough and difficulty breathing. The constant irritation and inflammation damages the lungs over time and can eventually lead to lung disease, heart disease, and death.
Sweat

Besides mucus, the CFTR protein also regulates other substances the body makes, like sweat and digestive fluids. In people with cystic fibrosis, the defective CFTR protein causes their bodies to excrete too much salt in their sweat, which depletes salt levels in the rest of the body.
If you have cystic fibrosis, this can cause serious problems during exercise or when spending time in the heat. Sweating can quickly lower salt levels in the body to dangerous levels, leading to severe dehydration, fatigue, muscle cramps, and even heat stroke.
That's why people with cystic fibrosis have to take extra care to stay hydrated, especially during physical activity. Drinking fluids like sports drinks that contain salt and other electrolytes can help replenish your salt levels, too.
Liver

Bile is an important digestive fluid that's released into your stomach when you eat food. This bile is made by the liver and passed to the gallbladder, where it is stored until it's needed.
Cystic fibrosis affects the bile-producing cells in the liver, causing them to secrete bile that's thicker than the usual flowing fluid. This can clog up t{{cta('fa8abc2a-1e88-4fa3-82fd-1cb5b9ed43b2','justifycenter')}}he liver and gallbladder and cause obstructions in the ducts that carry the bile, leading to liver scarring, gallstones, and other complications.
Pancreas
The pancreas is a vital part of the gastrointestinal system because it secretes the digestive enzymes needed to break down food. Cystic fibrosis affects the cells that secrete the pancreatic enzymes, causing the secretions to become thick and clog up the pancreas.
When this happens, enzymes are unable to make it into the intestines where they are needed to digest food. As a result, it doesn't get broken down correctly and the body cannot absorb the nutrients from the food you eat.
These nutrients are necessary for the body to grow and function, and untreated cystic fibrosis leads to illness and malnutrition. That's why 90 percent of people with the disease have to take pancreatic enzyme supplements with every meal—to make up for their pancreas's inability to supply its own digestive enzymes to the digestive tract.
The pancreas is also responsible for producing insulin, the substance that controls the amount of sugar in your blood. Cystic fibrosis can cause cysts and blockages in the pancreas that cause scarring and damage to the cells that make insulin.
Cystic fibrosis gets its name from these cysts that can develop in the pancreas as a result of the disease. However, it is not a very common occurrence, and only about 15 percent of people with cystic fibrosis develop this condition.
Intestines

Cystic fibrosis affects fluids secreted in the intestines, causing constipation and sometimes intestinal blockages. This happens because there is not enough water in the stool, and it can cause a serious condition in newborn babies called meconium ileus, which requires surgery.
Furthermore, cystic fibrosis prevents the pancreas from delivering digestive enzymes to the gut, which can cause serious complications in the gastrointestinal tract. This makes people with cystic fibrosis prone to malnourishment and symptoms like cramping, bloating, and constipation caused by undigested food that the body is unable to break down.
In the most severe cases, people with cystic fibrosis can have severe intestinal obstructions that completely block the intestines. Rectal prolapse is another complication that can arise from constant straining due to constipation and unusual bowel movements.
Key Facts about Cystic Fibrosis
Cystic fibrosis is a relatively rare disease, affecting about 30,000 people in the United states and up to 100,000 people worldwide. Although there is no way to cure the disease, there are many treatments available that focus on improving the symptoms, lifespan, and quality of life for people living with cystic fibrosis.

There are at least 1,700 different genetic mutations that can cause cystic fibrosis, and perhaps even more that haven't been studied. However, the vast majority of cystic fibrosis cases are caused by a single type of mutation, known as the F508 mutation. The rest are caused by one of the other hundreds of possible mutations.
The disease is serious, progressive, and tragically shortens the lifespan of people who suffer from it. However, with modern therapies people with cystic fibrosis are able to live much longer, better lives than ever before in history.
Until the late 1900's, most people born with cystic fibrosis didn't survive past elementary school. However, with proper treatment and medication the current median lifespan for people with cystic fibrosis is about 40 years old, and some live much longer.
Most cases of the disease are caught very early, often in newborn babies. In fact, hospitals have been required to do genetic screening for all newborns since 2010 to look for signs of cystic fibrosis. It's been very successful, and now nearly 65 percent of cystic fibrosis cases are found through newborn screening.
Cystic Fibrosis Symptoms
Since the disease is genetic, people with cystic fibrosis are born with it and usually start showing symptoms very early on in life. It is important to catch and begin treating the disease as early as possible to prevent serious complications and death.
The most prevalent and noticeable symptoms are coughing, phlegm, and shortness of breath. Many people with cystic fibrosis also fail to thrive and grow as children, and have difficulty putting on weight.
Cystic fibrosis is very taxing on the body and causes people with the disease to burn more energy and have large appetites. However, because their intestines aren't able to absorb nutrients efficiently, people with the disease are prone to getting malnourished and have difficulty gaining weight despite eating plenty of food.
Early-life Symptoms of Cystic Fibrosis:
- Salty skin and sweat
- Huge appetite (or reduced appetite, in some cases)
- Weight loss or difficulty putting on weight (failure to thrive)
- Low energy and fatigue
- Difficulty breathing
- Chronic coughing or wheezing
- Unusual bowel movements, such as constipation, chronic diarrhea, or large, greasy or foul-smelling stools.
Later-life Symptoms of Cystic Fibrosis:
- Difficulty breathing, especially with exercise
- Frequent lung infections
- Chronic cough with phlegm or blood
- Rectal prolapse from excessive straining
- Growths (polyps) in the nose and sinuses
- Clubbing (rounding and swelling) of the fingers
- Infertility
Causes of Cystic Fibrosis
Cystic fibrosis is an autosomal recessive genetic disease. That means that the only way you can get cystic fibrosis is if you are born with two defective copies of the CFTR gene; one inherited from each parent.

As we've explained, cystic fibrosis is a heritable disease that you have from birth, caused by mutated CFTR genes. There are around 1,700 different DNA mutations that can cause cystic fibrosis, but all of them lead to a defective CFTR protein that causes the body to secrete thickened mucus in the organs and airways.
Every person has two different versions of every gene, including the CFTR gene. Cystic fibrosis can be passed on to a child when two parents, who each have at least one defective CFTR gene, have a baby.
If the baby inherits one mutated gene and one normal gene from its parents, it would be a carrier of the disease, but not have any of the symptoms. Carriers can pass on the disease to to their own children, but only if they have a child with someone who is also a carrier.
When two parents who are carriers for cystic fibrosis have a baby, there is a 50% chance that the baby will receive one defective copy of the gene and be a carrier itself. There's a 25% chance the baby will inherit two defective copies of the gene and get cystic fibrosis.
Newborn babies are routinely given a genetic screening to test for the most common cystic fibrosis mutations. This test also catches many people who are carriers. However, since there are nearly two thousand different genetic mutations that can cause cystic fibrosis and possibly more undiscovered, genetic screening misses some babies who carry one defective copy of the gene.

Many doctors recommend that people who are carriers of cystic fibrosis and their partners get genetic screening and counseling if they plan to have a child.
Complications with Cystic Fibrosis
Infertility
The thickened mucus and other secretions caused by cystic fibrosis can lead to infertility in both males and females. That's because the fluids in the reproductive system are thickened and prone to causing blockages.
In males, thick secretions block the ability of sperm to pass through the testes, leaving men with cystic fibrosis unable to have children. Up to ninety percent of males with the disease are infertile.
In the female reproductive system, thickened mucus in the cervix can block sperm from passing into the uterus and fertilizing the egg, causing infertility. However, infertility is much less common in females than in males.
Liver Disease
In rare cases, thickened secretions caused by cystic fibrosis can cause bile to get stuck in the bile ducts in the liver. This causes an obstruction that leads to inflammation and scarring (cirrhosis).

Only about 5-10 percent of people with cystic fibrosis develop liver disease, and it is most often diagnosed before the age of 15. Still, doctors often recommend yearly liver screenings for people with cystic fibrosis to check for scarring and inflammation.
If you have cystic fibrosis, you can improve your chances of keeping a healthy liver by taking your medication properly, practicing good nutritional habits, and avoiding excessive alcohol use.
Pulmonary Hypertension and Heart Disease
Long-term respiratory issues can cause increased pressure in the lungs that compresses the surrounding blood vessels. Over time, this can cause pulmonary hypertension, a condition in which the pulmonary (lung) arteries get constricted, raising blood pressure in the lungs.

This puts back-pressure on the heart and forces it to work harder whenever it pumps blood through the vessels in the lungs. This extra strain causes the right ventricle (bottom-right chamber of the heart) to become enlarged over time.
This greatly weakens the heart and can cause edema (swollen legs and abdomen) and shortness of breath. Eventually, pulmonary hypertension and the associated enlarging of the right side of the heart can lead to right-sided heart failure (cor pulmonale) and death.
Gallbladder Disease
The gallbladder is responsible for collecting and storing the bile secreted by the liver. In people with cystic fibrosis, the bile can become so thick that it blocks the duct that leads out of the gallbladder to the intestine.
In most cases, this doesn't cause any discomfort or symptoms, but it sometimes causes gallstones to form. This occurs in about 10 percent of patients with cystic fibrosis and is usually treated with surgery to remove the gallbladder.
Pancreatitis
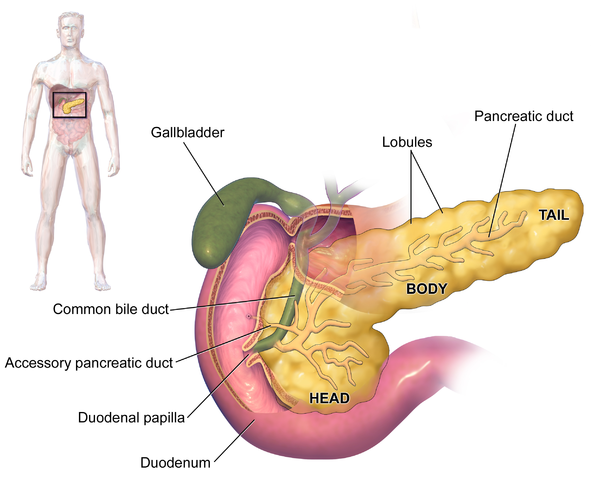
Both the gallbladder and the pancreas share a single duct that carries bile and pancreatic enzymes to the intestines. If cystic fibrosis causes gallstones to develop in the gallbladder, the stones can move up into the shared duct and block both bile and pancreatic secretions from getting through.
This can cause painful swelling and severe irritation in the pancreas, a serious condition known as pancreatitis. This can be treated with surgery to remove the gallstones or treatment in a hospital to reduce pain and discomfort until the inflammation subsides.
Bacterial Overgrowth
People with cystic fibrosis tend to develop frequent infections, especially lung infections, that require frequent courses of antibiotics. Over time, having to take antibiotics so often can kill off important, needed bacteria in the intestines.
This causes a bacteria imbalance that allows harmful bacteria to multiply. This can cause a number of uncomfortable symptoms, including nausea, gas, bloating, and diarrhea.
Bacterial overgrowth is often treated with a different kind of antibiotic that restores a healthy balance of bacteria in the gut. It selectively kills off harmful bacteria so that beneficial bacteria can thrive and flourish once more.
Diabetes and Hyperglycemia
Besides producing digestive enzymes, the pancreas has another very important purpose: secreting insulin into the bloodstream. Insulin is responsible for moving sugar out of the bloodstream and into the body's cells so it can be used as energy, and healthy insulin production is very important for overall health.

In rare cases, cystic fibrosis can cause cysts and scarring in the pancreas that destroys the cells that make insulin. Since the pancreas is the only organ that can make insulin, pancreatic scarring can severely reduce the amount of insulin in the blood.
This causes high blood sugar (hyperglycemia), which causes symptoms like dry mouth, fatigue, and frequent urination. Over time, insulin levels continue to decrease and blood sugar rises, eventually leading to diabetes.
Unlike type 2 diabetes, cystic fibrosis-related diabetes is not caused by diet or lifestyle habits, and is difficult to prevent. Fortunately, only about 15 percent of people with cystic fibrosis develop diabetes.
Gastric Paresis
Gastric Paresis occurs when food takes too long to empty out of the stomach, causing nausea, bloating, and discomfort after you eat. If you have pancreas scarring and high blood sugar, it can make the problem even worse.
Doctors often recommend that people with cystic fibrosis eat smaller, more frequent meals throughout the day to alleviate symptoms. This makes sure that you don't fill up your stomach too much, and allows it to empty more quickly and efficiently.
Delayed Puberty

Because of the digestive issues associated with cystic fibrosis, many children with the disease grow up with low body weights and nutrient deficiencies. In females, low weight and malnutrition can delay the onset of puberty.
Bronchiectasis
Bronchiectasis is a lung condition where the airways become enlarged and filled with mucus. This occurs often in people with cystic fibrosis as a result of chronic respiratory infections that inflame the airways and fill them with mucus, stretching them and causing permanent enlargement.

This makes the airways even more prone to infection in a self-perpetuating cycle that leads to more mucus, more enlargement, and more infections. This causes progressive damage to the lungs and airways over time, making it more and more difficult to breathe.
Antibiotics, expectorants, and bronchodilators are often prescribed to help treat bronchiectasis and make it easier to clear mucus from the airways. Practicing airway clearance techniques like chest percussion or postural drainage can help reduce symptoms and damage to the airways. Many patients with advanced bronciectasis require supplemental oxygen therapy as their lung function declines.
Pneumothorax

Pneumothorax is a medical term for the partial collapse of a lung. Anywhere from 5-20 percent of people with cystic fibrosis will develop a collapsed lung throughout their lifetime.
Pneumothorax is a life-threatening condition and happens when air leaks out of the lungs and fills the space around them, putting pressure on the lungs, heart, and blood vessels. This leaves less room for the lungs to expand, causing shortness of breath and pain when inhaling.
Risk Factors for Cystic Fibrosis
Since cystic fibrosis is caused by a heritable genetic mutation, you can only get cystic fibrosis if you inherit defective CFTR genes from both of your parents. Cystic fibrosis tends to occur most often in non-hispanic white people and is less common in people who belong to other racial and ethnic groups.
If you only inherit one defective gene, then you are a carrier. While carriers don't actually show any signs of cystic fibrosis themselves, they risk passing their defective gene on to their children.